物制藥有限公司")



搜索

新(xīn)聞中心
NEWS
資訊分(fēn)類
側邊欄
發布時(shí)間(jiān):2020-03-11 00:00:00


新(xīn)聞詳情
一(yī)文看懂FDA cGMP 483解析
- 分(fēn)類:行業資訊
- 作(zuò)者:巍信
- 來(lái)源:東富龍系統部
- 發布時(shí)間(jiān):2016-11-14 12:00
- 訪問量:
【概要描述】摘要FDA483報(bào)告,也(yě)稱現(xiàn)場(chǎng)檢查報(bào)告,是美國FDA檢查人(rén)員(yuán)根據美國現(xiàn)行法律法規,對企業進行現(xiàn)場(chǎng)檢查,對不符合cGMP的地方進行總結并發給企業。企業需立即針對不符合項進行整改,并在收到483報(bào)告後15個(gè)工作(zuò)日内向FDA相關(guān)部門回複483中不符合項的整改情況。若FDA認定企業的答(dá)複妥當,并符合現(xiàn)行法律法規的要求,那麽企業就(jiù)避免收到FDA警告信。FDA每個(gè)财政年

一(yī)文看懂FDA cGMP 483解析
【概要描述】摘要FDA483報(bào)告,也(yě)稱現(xiàn)場(chǎng)檢查報(bào)告,是美國FDA檢查人(rén)員(yuán)根據美國現(xiàn)行法律法規,對企業進行現(xiàn)場(chǎng)檢查,對不符合cGMP的地方進行總結并發給企業。企業需立即針對不符合項進行整改,并在收到483報(bào)告後15個(gè)工作(zuò)日内向FDA相關(guān)部門回複483中不符合項的整改情況。若FDA認定企業的答(dá)複妥當,并符合現(xiàn)行法律法規的要求,那麽企業就(jiù)避免收到FDA警告信。FDA每個(gè)财政年
- 分(fēn)類:行業資訊
- 作(zuò)者:巍信
- 來(lái)源:東富龍系統部
- 發布時(shí)間(jiān):2016-11-14 12:00
- 訪問量:
詳情
摘要
FDA483報(bào)告,也(yě)稱現(xiàn)場(chǎng)檢查報(bào)告,是美國FDA檢查人(rén)員(yuán)根據美國現(xiàn)行法律法規,對企業進行現(xiàn)場(chǎng)檢查,對不符合cGMP的地方進行總結并發給企業。企業需立即針對不符合項進行整改,并在收到483報(bào)告後15個(gè)工作(zuò)日内向FDA相關(guān)部門回複483中不符合項的整改情況。若FDA認定企業的答(dá)複妥當,并符合現(xiàn)行法律法規的要求,那麽企業就(jiù)避免收到FDA警告信。FDA每個(gè)财政年都會統計、總結并發布483表格,其在官網上(shàng)公布。本文通過對FDA官網發布的2010年~2015年483數據進行簡要分(fēn)析,并着重對FDA cGMP 483進行說(shuō)明和簡要分(fēn)析,旨在為(wèi)制藥企業完善内部管理(lǐ)體(tǐ)系提供一(yī)定的借鑒作(zuò)用。
1數據概覽
1、1483數據概況
FDA,中文全稱為(wèi)食品藥品監督管理(lǐ)局,其監管的範圍覆蓋了(le)食品、各種類型的藥品、醫(yī)療器(qì)械等行業,表1列出了(le)2010~2015财政年各個(gè)領域的檢查總結情況。
表1 2010~2015财政年發布的483份數
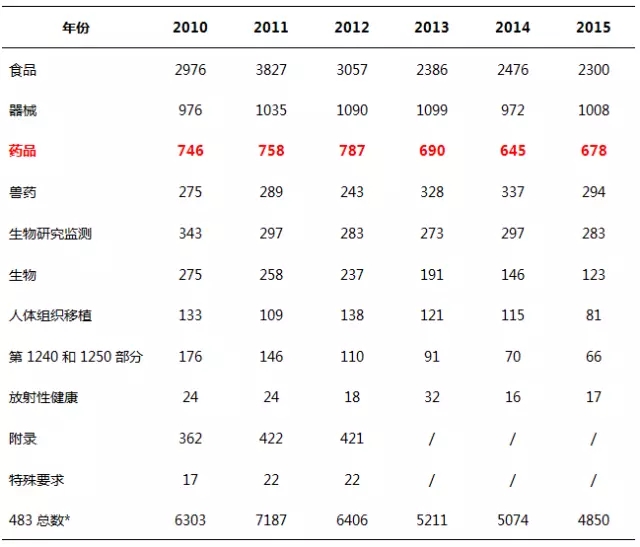
注:*以上(shàng)表格并不代表财政年發布的所有483份數,因為(wèi)部分(fēn)483為(wèi)手寫版,未包含在該表格中。
縱觀各個(gè)領域的483數據,食品483比重最大,約占總數的一(yī)半。藥品483比重從2010至2015财政年分(fēn)别為(wèi)11.9%、10.5%、12.3%、13.2%、12.7%、14.0%。表2為(wèi)2010~2015财政年藥品483數量情況。
表2 2010~2015财政年藥品483數量
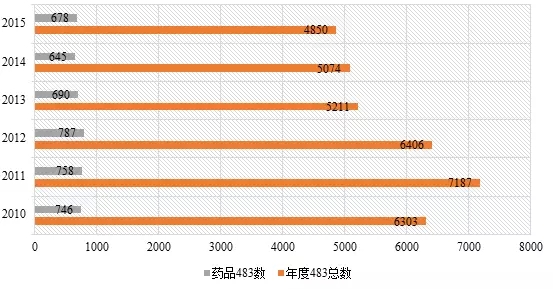
2、藥品483概況
藥品483涉及FDA頒發的聯邦法規第21章藥品相關(guān)法規和法律,包括成品藥良好(hǎo)(hǎo)生(shēng)産管理(lǐ)規範、PET藥品良好(hǎo)(hǎo)生(shēng)産管理(lǐ)規範、新(xīn)藥、聯邦食品藥品化(huà)妝品法律等。表3列出了(le)2010~2015财政年各法規出現(xiàn)不符合項的頻次。
表3 2010~2015财政年各法規出現(xiàn)不符合項的頻次
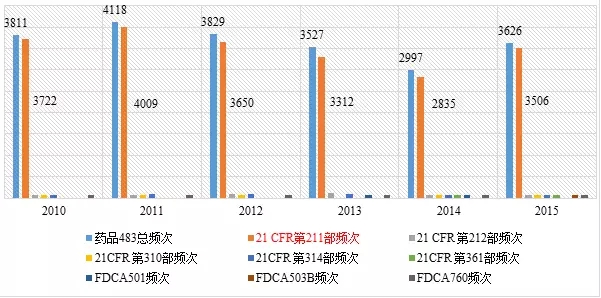
其中,21 CFR第211部是成品藥良好(hǎo)(hǎo)生(shēng)産管理(lǐ)規範(現(xiàn)行版本生(shēng)效日期為(wèi)2014年4月(yuè)(yuè)1日),21 CFR第212部是PET藥品良好(hǎo)(hǎo)生(shēng)産管理(lǐ)規範(現(xiàn)行版本生(shēng)效日期為(wèi)2014年4月(yuè)(yuè)1日),21 CFR第310部是新(xīn)藥(現(xiàn)行版本生(shēng)效日期為(wèi)2014年4月(yuè)(yuè)1日),21CFR第314部是新(xīn)藥上(shàng)市(shì)FDA批準申請(現(xiàn)行版本生(shēng)效日期為(wèi)2014年4月(yuè)(yuè)1日);21CFR第361部為(wèi)認為(wèi)安全、有效、未貼假商(shāng)标的處方藥:調查用藥(現(xiàn)行版本生(shēng)效日期為(wèi)2014年4月(yuè)(yuè)1日);FDCA501:聯邦食品、藥品和化(huà)妝品法501;FDCA503:聯邦食品、藥品和化(huà)妝品法503;FDCA760:聯邦食品、藥品和化(huà)妝品法760。
從表3的數據可知,21CFR第211部收到483報(bào)告的頻次最多,分(fēn)别占藥品483總頻次97.7%、97.4%、95.3%、93.9%、94.6%、96.7%。而21CFR第211部是美國FDA對藥品生(shēng)産的基本要求,是FDA檢查人(rén)員(yuán)檢查藥企遵循的重要準則。
3、FDA cGMP 483數據概況
FDA cGMP即21CFR第211部。表4按照六大體(tǐ)系展示不符合項的分(fēn)布情況。
表4藥品cGMP483分(fēn)布(按照六大體(tǐ)系劃分(fēn))
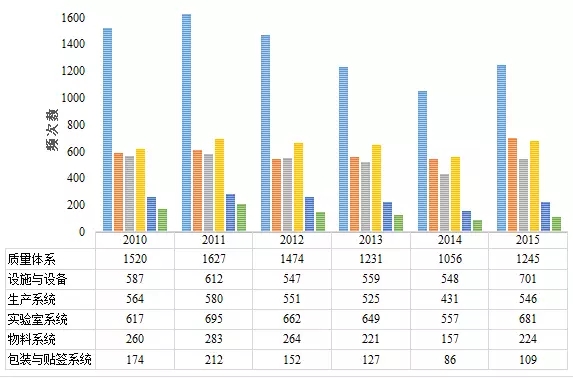
表4數據顯示,在六大體(tǐ)系中,質量體(tǐ)系出現(xiàn)不符合項的次數最多,大約占cGMP不符合項總數2/5;實驗室檢驗系統次之,比重大約為(wèi)cGMP不符合項總數1/6;廠房(fáng)設施與設備系統和生(shēng)産系統基本并列第三,約占cGMP不符合項總數1/7。
2六大體(tǐ)系483情況
1、質量體(tǐ)系483情況
根據表4的數據計算(suàn),2010~2015财政年質量體(tǐ)系出現(xiàn)不符合項占cGMP不符合項總數分(fēn)别為(wèi)40.8%、40.6%、40.4%、37.2%、37.2%、35.5%,逐年減少。表5彙總了(le)質量體(tǐ)系出現(xiàn)缺陷的部分(fēn)情況。
表5 質量體(tǐ)系不符合項部分(fēn)彙總
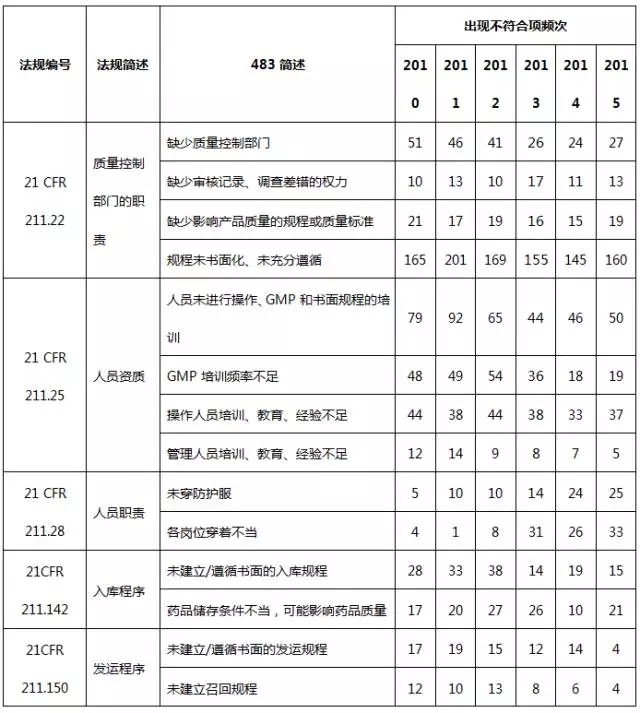
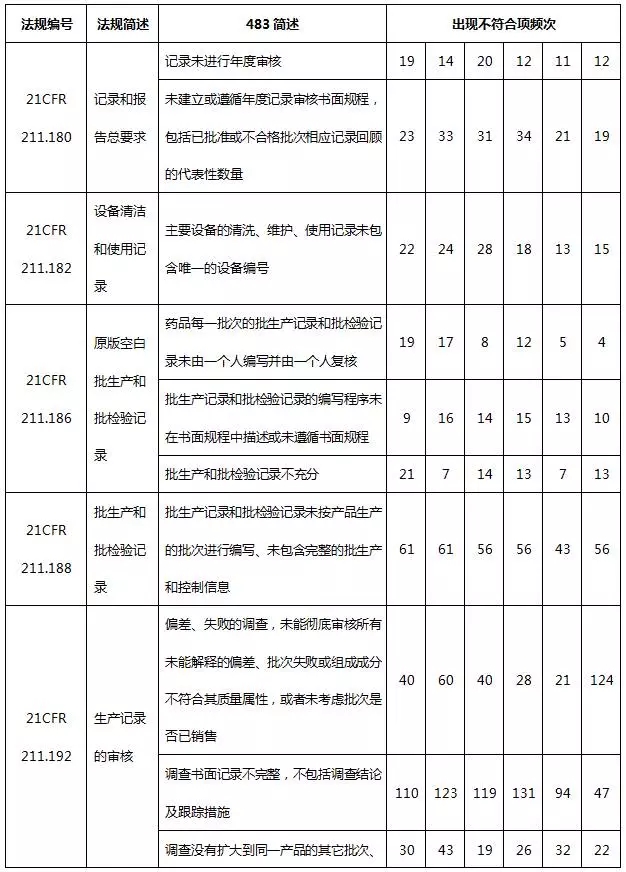
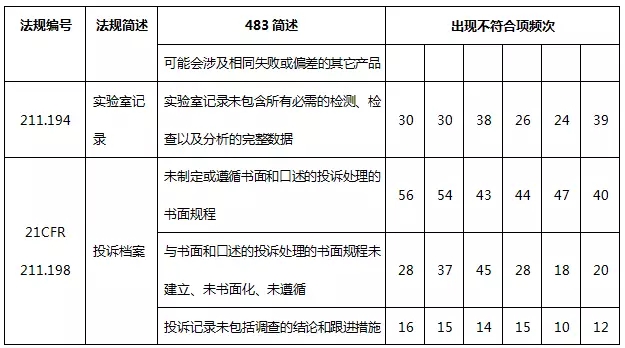
質量管理(lǐ)體(tǐ)系出現(xiàn)很多不合規的現(xiàn)象,主要表現(xiàn)于質量體(tǐ)系不健全,特别是文件體(tǐ)系,缺乏書面的質量管理(lǐ)規程或沒有充分(fēn)遵守已經制訂的規程,包括QC/QA管理(lǐ)規程、員(yuán)工培訓規程、入庫規程、發運規程以及各種記錄。另外,人(rén)員(yuán)教育、培訓及實際經驗缺乏也(yě)是常見的缺陷項。
制藥企業的質量體(tǐ)系水平在一(yī)定程度上(shàng)體(tǐ)現(xiàn)了(le)企業的質量文化(huà)水平,實際上(shàng)質量體(tǐ)系缺陷是一(yī)個(gè)國際性問題,其不僅出現(xiàn)在FDA 483中,在EudraGMDP不符合項報(bào)告中也(yě)很常見。制藥企業應該提高質量文化(huà)的建設,建立完善的質量體(tǐ)系,并定期評估質量體(tǐ)系運行的有效性。
2、實驗室檢驗系統483
2010~2015财政年實驗室檢驗系統不符合項出現(xiàn)概率分(fēn)别占16.6%、17.3%、18.1%、19.6%、19.6%、19.4%。以下(xià)就(jiù)實驗室檢驗系統不符合項進行部分(fēn)彙總。
表6 實驗室檢驗系統不符合項部分(fēn)彙總
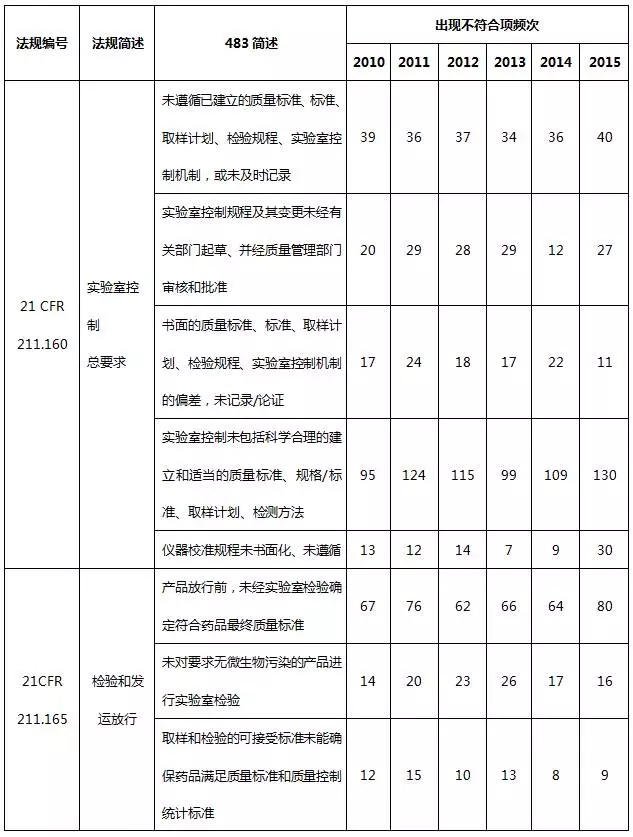
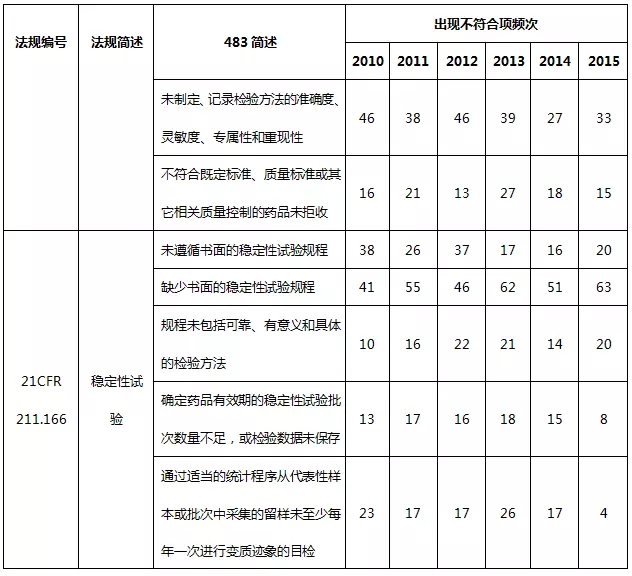
實驗室檢驗系統缺陷主要體(tǐ)現(xiàn)在質量标準、取樣計劃、檢驗規程、實驗室控制機制相關(guān)規程未建立、未充分(fēn)遵循以及未及時(shí)記錄,特别是實驗室控制規程,包括實驗室數據完整性、真實性、準确性,是頻發的不合規問題。當下(xià)制藥行業再次熱議(yì)數據完整性(CFDA已正式更名為(wèi)數據可靠性)這(zhè)一(yī)話(huà)題,不管是手工記錄還是電子(zǐ)記錄,均應以誠信為(wèi)基石。制藥企業可通過完善數據與記錄管理(lǐ)的技術基礎、完善記錄管理(lǐ)程序、完善記錄文件、建立計算(suàn)機化(huà)系統管理(lǐ)系統或高素質的員(yuán)工培訓來(lái)确保數據的完整性。
3、廠房(fáng)設施與設備系統483情況
2010~2015财政年廠房(fáng)設施與設備系統不符合項出現(xiàn)概率分(fēn)别占15.8%、15.3%、15.0%、16.9%、19.3%、20.0%。表7就(jiù)廠房(fáng)設施與設備系統不符合項進行部分(fēn)彙總。
表7 廠房(fáng)設施與設備系統不符合項部分(fēn)彙總
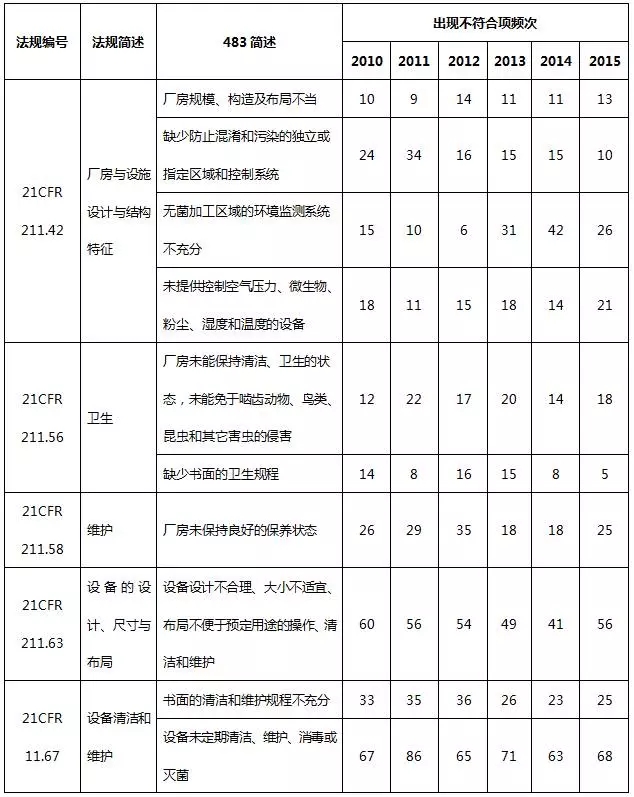
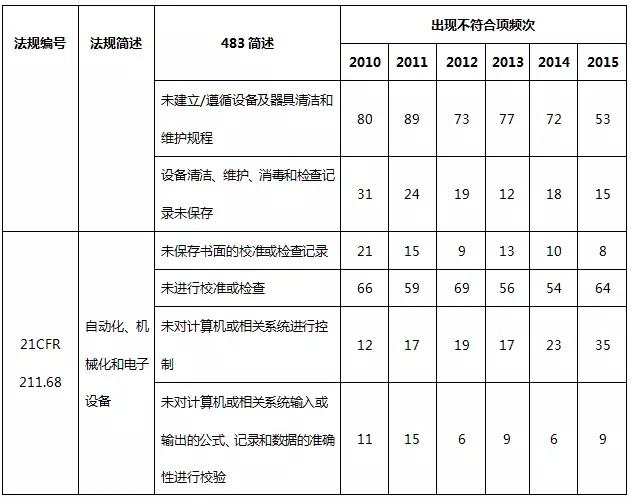
在廠房(fáng)設施與設備系統中,預防交叉污染、混淆和差錯不充分(fēn),設計、布局不合理(lǐ),不利于操作(zuò)、清潔和維護是主要的缺陷項目。自動化(huà)設備或儀器(qì)沒有進行準确性确認、校(xiào)準或控制,也(yě)時(shí)常被發現(xiàn)。
交叉污染是GMP的三大核心之一(yī),制藥企業應詳細評估車間(jiān)布局的可行性,并進行嚴格的驗證活動,避免挑戰法規的要求。自動化(huà)設備則應按照相關(guān)法規進行驗證,如(rú)CFDA最新(xīn)頒布的GMP附錄,計算(suàn)機化(huà)系統。
4、生(shēng)産系統483情況
2010~2015财政年生(shēng)産系統不符合項出現(xiàn)概率分(fēn)别占15.2%、14.5%、15.1%、15.9%、15.2%、15.6%。以下(xià)就(jiù)生(shēng)産系統不符合項進行部分(fēn)列表。
表8 生(shēng)産系統不符合項部分(fēn)彙總
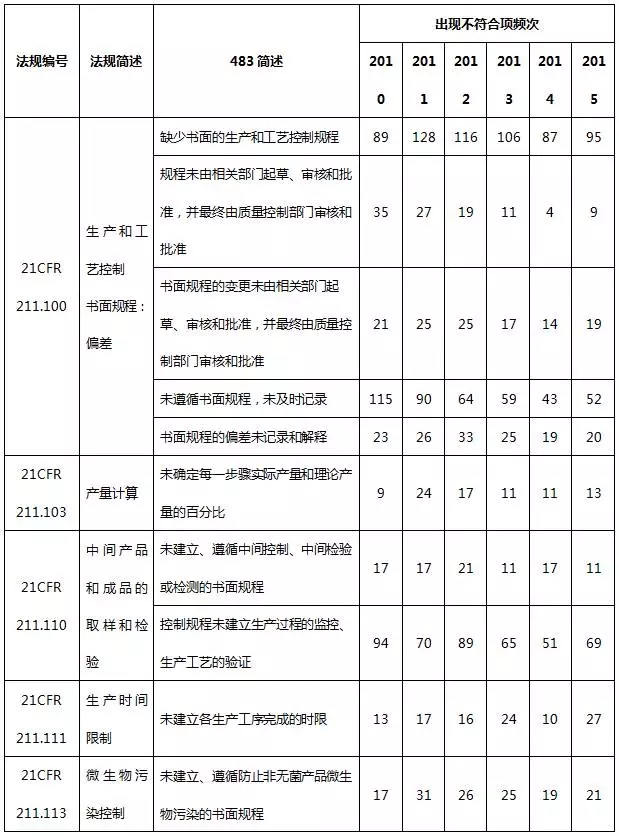
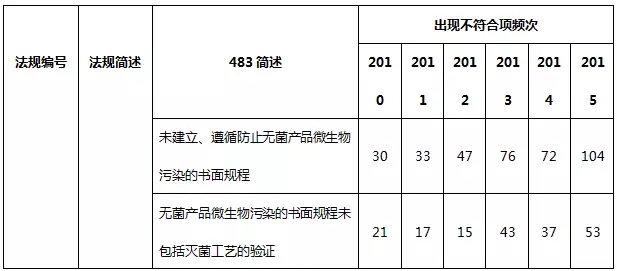
生(shēng)産系統常見的不合規項主要是缺少生(shēng)産和工藝控制、微生(shēng)物污染控制書面規程、未遵循書面規程、未及時(shí)記錄,未對每一(yī)步驟進行産量計算(suàn)以及未進行滅菌工藝驗證。值得一(yī)提的是,國内制藥企業通常會忽略除熱原工藝驗證,這(zhè)在FDA認證中将構為(wèi)嚴重缺陷。工藝驗證按照2011版FDA行業指南(nán)——工藝驗證的一(yī)般原則和規範提供的方法進行,其應貫穿藥品的整個(gè)生(shēng)命周期,包括藥物研發階段,一(yī)直到産品退市(shì)。
5、物料系統483情況
2010~2015财政年物料系統不符合項出現(xiàn)概率分(fēn)别占7.0%、7.1%、7.2%、6.7%、5.5%、6.4%。以下(xià)就(jiù)物料系統不符合項進行部分(fēn)列表。
表9 物料系統不符合項部分(fēn)彙總
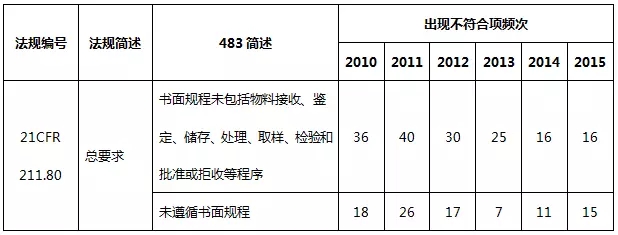
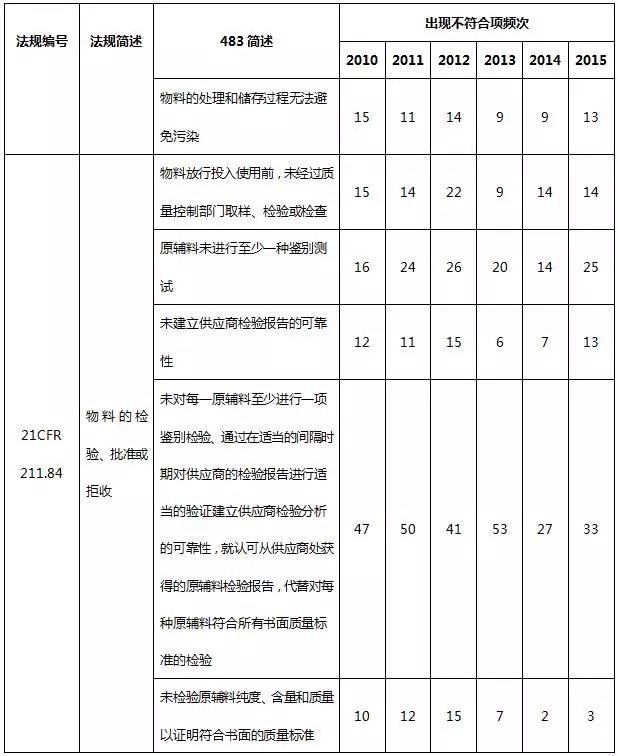
物料系統出現(xiàn)違規項目主要是書面規程不足、物料檢驗過分(fēn)依賴供應商(shāng)檢驗報(bào)告而未進行檢驗。從嚴格意義上(shàng)來(lái)說(shuō),表9列舉的缺陷項屬于質量體(tǐ)系的一(yī)部分(fēn),這(zhè)就(jiù)需要制藥企業提高質量的意識,從各個(gè)環節嚴格把控藥品生(shēng)産的質量。
6、包裝和貼簽系統483情況
2010~2015财政年包裝和貼簽系統不符合項出現(xiàn)概率分(fēn)别占4.7%、5.3%、4.2%、3.8%、3.0%、3.1%。表10彙總了(le)包裝和貼簽系統出現(xiàn)不符合項的部分(fēn)情況。
表10包裝和貼簽系統不符合項部分(fēn)彙總
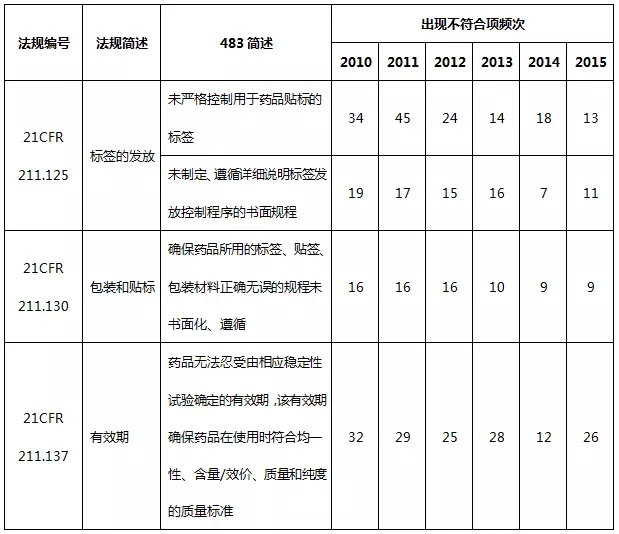
包裝和貼簽系統依舊存在規程未書面化(huà)或沒有充分(fēn)遵循的情況,此外有效期的确定不夠科學。同樣地,包裝和貼簽系統的缺陷項在一(yī)定程度上(shàng)也(yě)體(tǐ)現(xiàn)為(wèi)質量管理(lǐ)體(tǐ)系的不足。
3結束語
随着國内制藥企業水平的不斷提高,不少企業欲向歐美市(shì)場(chǎng)進軍,通過FDA cGMP是最低(dī)門檻,本文信息僅作(zuò)為(wèi)制藥企業對照現(xiàn)有的質量保證體(tǐ)系是否存在違反FDAcGMP的參考。特别是在規程書面化(huà)、完整化(huà)以及充分(fēn)遵循化(huà)方面,不管是在質量體(tǐ)系還是包裝貼簽體(tǐ)系,對出現(xiàn)的違規現(xiàn)象應該引起廣大制藥企業的深思和警醒。
掃二維碼用手機看
上(shàng)一(yī)個(gè):
無
下(xià)一(yī)個(gè):
CFDA放(fàng)大招,自查之後将鐵血飛檢!
上(shàng)一(yī)個(gè):
無
下(xià)一(yī)個(gè):
CFDA放(fàng)大招,自查之後将鐵血飛檢!
頁面版權所有 湖北億諾瑞生(shēng)物制藥有限公司 地 址:湖北省黃(huáng)梅縣小池鎮沿江路(lù)108号 證書編号:(鄂)-非經營性-2018-0070