物制藥有限公司")
搜索




新(xīn)聞中心
NEWS
資訊分(fēn)類
側邊欄
發布時(shí)間(jiān):2020-03-11 00:00:00


新(xīn)聞詳情
CFDA放(fàng)大招,自查之後将鐵血飛檢!
- 分(fēn)類:行業資訊
- 作(zuò)者:
- 來(lái)源:CFDA
- 發布時(shí)間(jiān):2016-11-14 12:00
- 訪問量:
【概要描述】總局辦公廳公開征求《關(guān)于開展藥品生(shēng)産工藝核對工作(zuò)的公告(征求意見稿)》的意見為(wèi)進一(yī)步規範藥品生(shēng)産工藝管理(lǐ),保障公衆用藥安全,食品藥品監管總局組織起草了(le)《關(guān)于開展藥品生(shēng)産工藝核對工作(zuò)的公告(征求意見稿)》,現(xiàn)向社會公開征求意見。請将修改意見于2016年9月(yuè)(yuè)10日前通過電子(zǐ)郵件反饋至食品藥品監管總局藥品化(huà)妝品注冊管理(lǐ)司。聯系人(rén):薛彥久電子(zǐ)郵箱:yhzcszhc@

CFDA放(fàng)大招,自查之後将鐵血飛檢!
【概要描述】總局辦公廳公開征求《關(guān)于開展藥品生(shēng)産工藝核對工作(zuò)的公告(征求意見稿)》的意見為(wèi)進一(yī)步規範藥品生(shēng)産工藝管理(lǐ),保障公衆用藥安全,食品藥品監管總局組織起草了(le)《關(guān)于開展藥品生(shēng)産工藝核對工作(zuò)的公告(征求意見稿)》,現(xiàn)向社會公開征求意見。請将修改意見于2016年9月(yuè)(yuè)10日前通過電子(zǐ)郵件反饋至食品藥品監管總局藥品化(huà)妝品注冊管理(lǐ)司。聯系人(rén):薛彥久電子(zǐ)郵箱:yhzcszhc@
- 分(fēn)類:行業資訊
- 作(zuò)者:
- 來(lái)源:CFDA
- 發布時(shí)間(jiān):2016-11-14 12:00
- 訪問量:
詳情
總局辦公廳公開征求《關(guān)于開展藥品生(shēng)産工藝核對工作(zuò)的公告(征求意見稿)》的意見
為(wèi)進一(yī)步規範藥品生(shēng)産工藝管理(lǐ),保障公衆用藥安全,食品藥品監管總局組織起草了(le)《關(guān)于開展藥品生(shēng)産工藝核對工作(zuò)的公告(征求意見稿)》,現(xiàn)向社會公開征求意見。請将修改意見于2016年9月(yuè)(yuè)10日前通過電子(zǐ)郵件反饋至食品藥品監管總局藥品化(huà)妝品注冊管理(lǐ)司。
聯系人(rén):薛彥久
電子(zǐ)郵箱:yhzcszhc@cfda.gov.cn
附件:關(guān)于開展藥品生(shēng)産工藝核對工作(zuò)的公告(征求意見稿)
食品藥品監管總局辦公廳
2016年8月(yuè)(yuè)9日附件:關(guān)于開展藥品生(shēng)産工藝核對工作(zuò)的公告(征求意見稿)
藥品生(shēng)産工藝是持續穩定地生(shēng)産出合格藥品的過程和方法,按照監管部門批準的生(shēng)産工藝組織生(shēng)産是保障藥品質量的前提。為(wèi)加強藥品生(shēng)産工藝管理(lǐ),原國家食品藥品監督管理(lǐ)局于2007年8月(yuè)(yuè)部署開展了(le)注射劑類藥品生(shēng)産工藝和處方核查工作(zuò),各省局對企業申報(bào)登記的生(shēng)産工藝等相關(guān)資料進行了(le)審查,初步建立了(le)注射劑生(shēng)産工藝等資料檔案。2007年10月(yuè)(yuè)修訂實施的《藥品注冊管理(lǐ)辦法》規定批準藥品上(shàng)市(shì)前應進行生(shēng)産現(xiàn)場(chǎng)檢查,保障了(le)技術審評部門核定的生(shēng)産工藝的可行性,此後批準上(shàng)市(shì)的絕大多數藥品的實際生(shēng)産工藝與批準的生(shēng)産工藝是一(yī)緻的。近年來(lái),食品藥品監管部門在監督檢查中發現(xiàn)仍有部分(fēn)2007年前批準上(shàng)市(shì)的品種未按照批準的生(shēng)産工藝組織生(shēng)産、改變生(shēng)産工藝不按規定研究和申報(bào)。為(wèi)此,總局決定開展藥品生(shēng)産工藝核對工作(zuò)。現(xiàn)就(jiù)有關(guān)事(shì)項公告如(rú)下(xià):
一(yī)、藥品生(shēng)産企業承擔藥品質量安全的主體(tǐ)責任,必須嚴格按照食品藥品監管部門批準的生(shēng)産工藝組織生(shēng)産。藥品生(shēng)産企業改變已批準的生(shēng)産工藝,必須經過充分(fēn)的研究和驗證,并按照《藥品注冊管理(lǐ)辦法》的有關(guān)規定提交藥品注冊補充申請。
二、自本公告發布之日起,藥品生(shēng)産企業應對每個(gè)批準上(shàng)市(shì)藥品的生(shēng)産工藝(中藥為(wèi)制法,下(xià)同)開展自查,排除質量安全隐患。
三、自查内容為(wèi)藥品的實際生(shēng)産工藝與報(bào)經食品藥品監管部門批準的生(shēng)産工藝是否一(yī)緻。食品藥品監管部門批準的生(shēng)産工藝包括審批藥品生(shēng)産申請時(shí)批準的生(shēng)産工藝及審批相關(guān)補充申請時(shí)批準的生(shēng)産工藝。
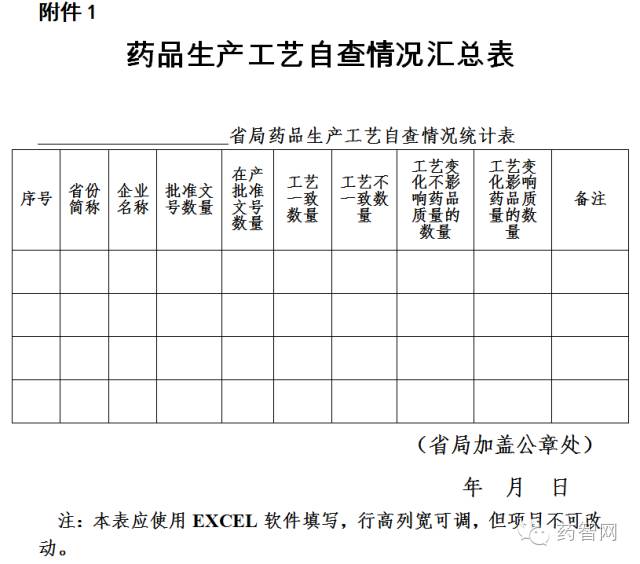
四、藥品生(shēng)産企業應于2016年10月(yuè)(yuè)1日前完成自查并将自查情況報(bào)所在地省級食品藥品監管部門。省級食品藥品監管部門應對企業自查情況進行彙總,填寫自查情況彙總表(附件1)并于2016年11月(yuè)(yuè)1日前上(shàng)報(bào)食品藥品監管總局。
五、藥品生(shēng)産企業根據自查結果,應分(fēn)别采取以下(xià)處理(lǐ)措施:
(一(yī))實際生(shēng)産工藝與批準生(shēng)産工藝一(yī)緻、能(néng)夠保證藥品質量的,藥品生(shēng)産企業應将自查情況報(bào)告與藥品生(shēng)産工藝等資料一(yī)并歸檔,作(zuò)為(wèi)監管部門開展日常監管、現(xiàn)場(chǎng)核查的備查資料。
(二)實際生(shēng)産工藝與批準生(shēng)産工藝不一(yī)緻的,相關(guān)藥品生(shēng)産企業應按照《藥品生(shēng)産質量管理(lǐ)規範》《藥品注冊管理(lǐ)辦法》補充申請事(shì)項的相關(guān)要求以及《已上(shàng)市(shì)中藥變更研究技術指導原則(一(yī))》《已上(shàng)市(shì)化(huà)學藥品變更研究的技術指導原則(一(yī))》《生(shēng)物制品生(shēng)産工藝過程變更管理(lǐ)技術指導原則》等相關(guān)技術要求開展充分(fēn)的研究驗證。
經研究驗證,生(shēng)産工藝變化(huà)對藥品質量不産生(shēng)影響的,藥品生(shēng)産企業應按照《藥品注冊管理(lǐ)辦法》附件4第18項提出補充申請,申報(bào)資料要求見附件2。省級食品藥品監管部門于受理(lǐ)後5日内将申報(bào)資料送交國家食品藥品監督管理(lǐ)總局藥品審評中心(以下(xià)簡稱“藥審中心”)。藥審中心依據《藥品注冊管理(lǐ)辦法》開展技術審評,必要時(shí)可以要求申請人(rén)補充資料,所需時(shí)間(jiān)不計入技術審評時(shí)限。國家食品藥品監督管理(lǐ)總局依據《藥品注冊管理(lǐ)辦法》作(zuò)出審批決定。
經研究驗證,生(shēng)産工藝變化(huà)對藥品質量産生(shēng)影響的,企業應立即停産。藥品生(shēng)産企業應按照《藥品注冊管理(lǐ)辦法》附件4第7項提出“改變影響藥品質量的生(shēng)産工藝”補充申請。省級食品藥品監管部門于受理(lǐ)後5日内将申報(bào)資料送交藥審中心。藥審中心應組織專門審評力量、建立單獨審評通道,于收到申報(bào)資料後30日内完成技術審評,必要時(shí)可以要求申請人(rén)補充資料,所需時(shí)間(jiān)不計入技術審評時(shí)限。國家食品藥品監督管理(lǐ)總局應在5日内完成行政審批。補充申請獲批後,藥品生(shēng)産企業方可繼續生(shēng)産。
藥品生(shēng)産企業應于2017年6月(yuè)(yuè)30日前完成在産品種生(shēng)産工藝的研究驗證、提交補充申請等相關(guān)工作(zuò),其他暫不生(shēng)産品種應于2017年12月(yuè)(yuè)31日前完成上(shàng)述工作(zuò);未按時(shí)完成的,應停止生(shēng)産。
六、2016年11月(yuè)(yuè)1日起,國家食品藥品監督管理(lǐ)總局将組織專家對藥品生(shēng)産企業開展飛行檢查。檢查中發現(xiàn)實際生(shēng)産工藝與食品藥品監管部門批準的生(shēng)産工藝不一(yī)緻的,依據《中華人(rén)民(mín)共和國藥品管理(lǐ)法》第四十八條第二款的有關(guān)規定,其所生(shēng)産的藥品按假藥論處。藥監部門将依據《中華人(rén)民(mín)共和國藥品管理(lǐ)法》第七十四條的有關(guān)規定對涉事(shì)藥品生(shēng)産企業進行處罰,并向社會公開相關(guān)企業法定代表人(rén)和相關(guān)責任人(rén)員(yuán)。
七、發生(shēng)過影響藥品質量的生(shēng)産工藝變更,但(dàn)生(shēng)産企業能(néng)夠确保産品安全有效的,符合下(xià)列情形的,可以暫不停産,但(dàn)需要按本公告要求提出相關(guān)補充申請。
(一(yī))相關(guān)品種在《藥品注冊管理(lǐ)辦法》2007年修訂實施前已經發生(shēng)影響藥品質量的生(shēng)産工藝變更,此後一(yī)直正常生(shēng)産,生(shēng)産工藝穩定且未發現(xiàn)安全性和有效性問題的;
(二)相關(guān)品種發生(shēng)影響藥品質量的生(shēng)産工藝變更,變更後的生(shēng)産工藝屬于技術進步或創新(xīn)的。
八、本公告自發布之日起實施,進口藥品參照執行。
特此公告。
附件:1.藥品生(shēng)産工藝自查情況彙總表
2.申報(bào)資料要求(不影響藥品質量的生(shēng)産工藝變更)
3.藥品生(shēng)産工藝變更情況表附件2申報(bào)資料要求(不影響藥品質量的生(shēng)産工藝變更)
一(yī)、申報(bào)資料内容
(一(yī))申請表
國産藥品、進口藥品及港澳台醫(yī)藥産品分(fēn)别提供相應的注冊—(補充)申請表。
(二)藥品批準證明文件及其附件的複印件
包括藥品注冊批件及其附件、藥品注冊證、已取得的《藥品補充申請批件》及其附件、《審批意見通知件》、備案情況公示相關(guān)文件、尚處于審批期間(jiān)的相關(guān)補充申請的受理(lǐ)通知書/簽收單複印件。
進口藥品,應當提交其生(shēng)産國家或者地區藥品管理(lǐ)機構出具的允許藥品工藝變更的證明文件及其中文譯本。
(三)證明性文件
包括《藥品生(shēng)産許可證》及其變更記錄頁複印件、營業執照複印件、《藥品GMP證書》複印件。
(四)藥學研究資料
包括制劑的藥品處方、藥品生(shēng)産工藝及變更評估資料。
藥品處方應包括活性成分(fēn)或中藥藥味、輔料的種類和數量,寫明按1000制劑單位的理(lǐ)論處方量、上(shàng)市(shì)生(shēng)産批量,以及實際過量(減量)投料的情況和理(lǐ)由。輔料包括着色劑、防腐劑、香料、矯味劑、包衣劑(應明确規格型号)、空心膠囊、pH調節劑、制劑過程中去除的溶劑(如(rú)乙醇等,純化(huà)水除外)等。除了(le)pH調節劑和中藥處方中必須通過調節用量達到片重規格的特定輔料以外,均應注明輔料的具體(tǐ)處方量或經驗證的處方量範圍。
生(shēng)産工藝應包括工藝流程圖和工藝過程描述。原注冊申報(bào)的工藝過于簡單或關(guān)鍵信息不夠明确的,應當在備案申報(bào)資料中将關(guān)鍵信息補充完整。
工藝流程圖應明确物料及投料批量、各工藝步驟的具體(tǐ)操作(zuò)過程、關(guān)鍵工藝參數及生(shēng)産過程監測、中間(jiān)體(tǐ)質控、主要設備名稱及型号、潔淨背景等主要信息。
工藝過程描述應包括從物料準備到完成成品包裝的完整生(shēng)産過程,詳略程度應能(néng)使本專業的技術人(rén)員(yuán)根據申報(bào)的生(shēng)産工藝可以完整地重複生(shēng)産過程,并制得符合标準的産品;應明确每個(gè)單元操作(zuò),按單元操作(zuò)過程描述工藝,明确投料量或投料比、操作(zuò)流程、各主要工序的關(guān)鍵工藝參數及範圍、主要設備和材料類型、中間(jiān)過程的取樣和主要質量控制要求(包括中間(jiān)體(tǐ)檢驗的檢測項目及限度),并注明經驗證的生(shēng)産規模和收率範圍。生(shēng)産工藝中如(rú)用到特殊儀器(qì)設備、操作(zuò)方法、檢驗檢查方法及其他過程控制要求的,應明确闡述、說(shuō)明。
原料藥的生(shēng)産工藝描述應明确起始物料來(lái)源、規格和标準、批量、各步驟物料(包括起始物料、溶劑、催化(huà)劑、氣體(tǐ)等)用量/配比/濃度、加料順序、加料速度、溫度、壓力、真空度、時(shí)間(jiān)、攪拌速度、分(fēn)離純化(huà)柱的材質型号、精制方法和次數、中間(jiān)體(tǐ)質控要求、主要設備類型等。原批準工藝為(wèi)采用市(shì)售原料藥粗品或遊離酸/堿(除無機化(huà)合物或已上(shàng)市(shì)原料藥外)一(yī)步成鹽而精制的原料藥,應提供粗品或遊離酸/堿的供應商(shāng)、詳細生(shēng)産工藝和過程控制資料。
中藥提取應明确藥材(飲片、提取物)的來(lái)源、标準、預處理(lǐ)方法條件和工藝參數、提取方法及條件、溶媒的種類、用量、提取次數、提取溫度、時(shí)間(jiān)、提取液過濾的方法及條件、濃縮的方法及條件、濃縮過程允許的最長受熱時(shí)間(jiān)、濃縮液的相對密度、濃縮液或浸膏的得率範圍、濃縮液的貯存條件和期限、純化(huà)的方法及條件如(rú)溶劑濃度、相對密度、測定溫度、攪拌方法和條件、分(fēn)離純化(huà)柱的材質型号、精制方法和次數等、提取物幹燥的方法、溫度上(shàng)限、最長受熱時(shí)間(jiān)、真空度、收率範圍等。
制劑的生(shēng)産工藝描述應明确原輔料處理(lǐ)方式、粒度控制、投料量(投料比)、投料順序、制粒方法、過篩目數/篩網孔徑、攪拌速度/頻率、混合時(shí)間(jiān)、溶劑及粘合劑的配制濃度及用量、幹燥方式、幹燥溫度、幹燥時(shí)間(jiān)或其他終點控制參數、攪拌或混合方式、轉速/頻率、混合時(shí)間(jiān)、溫度、pH值、溶解時(shí)間(jiān)、氣體(tǐ)保護(如(rú)需充氮氣,充氮氣時(shí)間(jiān))、濾材種類、型号級别、規格、過濾方式、藥液的溫度與流速、凍幹曲線的參數設置、滅菌方法、滅菌櫃類型、裝載方式、滅菌時(shí)間(jiān)、設定溫度、壓力、F0值等。
需要開展藥理(lǐ)毒理(lǐ)或者臨床研究的,按照現(xiàn)行藥品注冊要求及程序開展相應研究工作(zuò)。
二、申報(bào)資料的提交
申請人(rén)應當提供上(shàng)述申報(bào)資料1套,并按資料項目編号順序整理(lǐ)。每項申報(bào)資料應設置封面和編号後單獨裝訂,封面加蓋申報(bào)單位公章。
藥品處方、生(shēng)産工藝應建立word格式電子(zǐ)文件,并壓縮成一(yī)個(gè)zip文件,命名為(wèi)“批準文号後9位代碼附件”。該電子(zǐ)文件應一(yī)并提交,并導入申請表附
附件3 藥品生(shēng)産工藝變更情況表
一(yī)、不影響藥品質量的生(shēng)産工藝變更
(一(yī))中藥I類和Ⅱ類變更
1.I類變更:此類變更不會引起藥用物質基礎的改變,對藥物的吸收、利用不會産生(shēng)明顯影響,不會引起安全性、有效性的明顯改變。如(rú)變更不含揮發性成分(fēn)、熱敏性成分(fēn)藥物的粉碎工藝(其粉碎粒度基本相同)、濃縮幹燥工藝或制粒工藝(縮短受熱時(shí)間(jiān)或降低(dī)受熱溫度)等,但(dàn)變更為(wèi)特殊的濃縮幹燥方法,如(rú)微波幹燥等方法,不屬于此類變更。
2.II類變更:此類變更對其藥用物質基礎或對藥物的吸收、利用有影響,但(dàn)變化(huà)不大,包括工藝過程中一(yī)些(xiē)工藝參數及工藝方法的改變,如(rú)變更含揮發性成分(fēn)、熱敏性成分(fēn)藥物的涉及受熱溫度、受熱時(shí)間(jiān)的工藝操作(zuò),應進行對比研究,如(rú)藥用物質變化(huà)不大,屬于Ⅱ類變更。
(二)化(huà)學藥品I類和Ⅱ類變更
1.變更原料藥的生(shēng)産工藝:
I類變更:
(1)變更試劑、起始原料的來(lái)源;
(2)提高試劑、起始原料、中間(jiān)體(tǐ)的質量标準;
II類變更:
(3)變更起始原料、溶劑、試劑、中間(jiān)體(tǐ)的質量标準;
2.變更制劑的生(shēng)産工藝:
I類變更:
(1)增加生(shēng)産過程質量控制方法或嚴格控制限度;
(2)片劑、膠囊、栓劑或陰道栓印記變更;
(3)普通或腸溶片劑、膠囊、栓劑或陰道栓的形狀、尺寸變更; II類變更:
(4)變更生(shēng)産設備;包括無菌制劑生(shēng)産中采用相同設計及操作(zuò)原理(lǐ)的設備替代另一(yī)種設備;非無菌制劑生(shēng)産中采用設計及操作(zuò)原理(lǐ)不同的設備替代另一(yī)種設備;改變半固體(tǐ)制劑生(shēng)産中混合設備類型,由高速剪切機變更為(wèi)低(dī)速剪切機,或相反變更。如(rú)涉及無菌産品時(shí),變更生(shēng)産設備不應降低(dī)産品的無菌保證水平。
(5)變更制劑生(shēng)産過程,包括口服固體(tǐ)制劑物料混合過程的混合時(shí)間(jiān)及混合速度等變更,包括半固體(tǐ)制劑混合過程中的混合速度、混合時(shí)間(jiān)、冷卻速度等生(shēng)産過程的變更,還包括半固體(tǐ)制劑水相與油相混合過程的變更。對于無菌制劑,這(zhè)種變更包括:①對采用終端滅菌工藝生(shēng)産的無菌制劑,取消中間(jiān)過程的濾過環節;②變更除菌過濾過程的濾過參數(包括流速、壓力、時(shí)間(jiān)、或體(tǐ)積,但(dàn)濾過材料和孔徑不變)等。此類變更不應引起制劑生(shēng)産工藝的根本性改變,不引起産品與體(tǐ)内吸收和療效有關(guān)的重要理(lǐ)化(huà)性質和指标的改變。無菌産品生(shēng)産過程變更應不降低(dī)産品的無菌保證水平。
(6)緩釋或控釋片劑、膠囊、栓劑或陰道栓形狀、尺寸變更,包括片劑、膠囊、栓劑或陰道栓形狀變化(huà),如(rú)圓形片變為(wèi)異形片(菱形等)等,但(dàn)制劑處方沒有改變。對于緩釋制劑/控釋制劑,制劑形狀與藥物釋放(fàng)行為(wèi)有一(yī)定關(guān)系,因此,外形變化(huà)在某些(xiē)時(shí)候對藥物釋放(fàng)行為(wèi)可能(néng)是有影響的,需注意對變更前後藥物釋放(fàng)行為(wèi)進行較為(wèi)充分(fēn)的比較研究。
(三)生(shēng)物制品Ⅲ類變更
1.菌毒種庫及細胞庫
工作(zuò)種子(zǐ)庫
2.生(shēng)産工藝 緩沖液
3.配制 稀釋劑(新(xīn)稀釋劑除外)
(四)其他經研究驗證和評估确認不影響藥品安全性、有效性和質量可控性的工藝變更。
二、影響藥品質量的生(shēng)産工藝變更
(一(yī))中藥Ⅲ類變更
此類變更會引起藥用物質基礎的明顯改變,或對藥物的吸收、利用可能(néng)産生(shēng)明顯影響,如(rú)工藝路(lù)線改變,包括藥材合并提取與分(fēn)開提取的改變、提取溶媒種類的改變;工藝方法改變,包括純化(huà)方法由醇沉改為(wèi)澄清劑處理(lǐ),減壓幹燥改為(wèi)微波幹燥等特殊幹燥方法,對藥物吸收利用有明顯影響的成型工藝方法改變等;工藝參數改變,包括醇沉工藝中醇沉含醇量的改變,提取次數的改變等。
(二)化(huà)學藥品Ⅲ類變更
1.變更原料藥的生(shēng)産工藝
(1)主要包括變更反應條件,變更某一(yī)步或幾步反應,甚至整個(gè)合成路(lù)線等,将原合成路(lù)線中的某中間(jiān)體(tǐ)作(zuò)為(wèi)起始原料的工藝變更等。
2.變更制劑的生(shēng)産工藝
(1)制劑生(shēng)産過程或生(shēng)産工藝發生(shēng)重大變化(huà)的,如(rú)口服固體(tǐ)制劑由濕法制粒改變為(wèi)幹法制粒,或相反變更;如(rú)生(shēng)産過程幹燥方法從烘箱幹燥變為(wèi)流化(huà)床幹燥或相反變更等。
(2)制劑生(shēng)産工藝變更可能(néng)影響制劑控釋或緩釋特性的,可能(néng)影響制劑(如(rú)吸入劑、噴霧劑)體(tǐ)内吸收的,或影響制劑其他特性(如(rú)藥物粒度)的。
(3)無菌生(shēng)産過程變更可能(néng)影響藥品無菌保證水平的,包括:①變更産品滅菌工藝,由除菌過濾滅菌工藝變更為(wèi)終端滅菌工藝;如(rú)終端滅菌工藝由殘存概率法變更為(wèi)過度殺滅法;從幹熱滅菌、輻射滅菌中的一(yī)種滅菌工藝變更為(wèi)另一(yī)種滅菌工藝等。②用不同操作(zuò)原理(lǐ)的滅菌櫃替代原滅菌櫃。③變更滅菌櫃的藥品裝載量和裝載方式,且超出原驗證的範疇的。④變更除菌過濾過程的濾材種類或孔徑。⑤使用不同容量的凍幹設備替代原凍幹設備,或增加不同容量的凍幹設備,新(xīn)的凍幹設備與原凍幹設備的操作(zuò)參數和總的生(shēng)産時(shí)間(jiān)有改變。
(三)生(shēng)物制品I類和Ⅱ類變更
1.主要原輔材料
I類變更:
(1)原材料或起始原材料
II類變更
(2)培養基或其主要成份
(3)關(guān)鍵原輔料的來(lái)源
(4)牛血清及胰酶等
2.菌毒種庫及細胞庫
I類變更:原始種子(zǐ)庫
II類變更:主代種子(zǐ)庫
3.生(shēng)産工藝
I類變更
(1)病毒滅活方法變更
II類變更
(2)減少或增加工藝步驟
(3)培養時(shí)間(jiān)變更
(4)分(fēn)離、純化(huà)方法變更
(5)參數變更
(6)生(shēng)産規模改變
4.配制
II類變更
(1)防腐劑
(2)佐劑(新(xīn)佐劑除外)
(3)賦型劑
(4)穩定劑
5.生(shēng)産設備
I類變更:
(1)主要生(shēng)産設備(如(rú)消毒、凍幹、分(fēn)裝、發酵罐血液制品生(shēng)産用離心機、壓濾機)
II類變更:
(2)一(yī)般生(shēng)産設備
(四)其他經研究驗證和評估認為(wèi)影響或可能(néng)影響藥品安全性、有效性和質量可控性的工藝變更。
掃二維碼用手機看
上(shàng)一(yī)個(gè):
一(yī)文看懂FDA cGMP 483解析
下(xià)一(yī)個(gè):
低(dī)分(fēn)子(zǐ)肝素産品升級,靜觀市(shì)場(chǎng)格局生(shēng)變
上(shàng)一(yī)個(gè):
一(yī)文看懂FDA cGMP 483解析
下(xià)一(yī)個(gè):
低(dī)分(fēn)子(zǐ)肝素産品升級,靜觀市(shì)場(chǎng)格局生(shēng)變
頁面版權所有 湖北億諾瑞生(shēng)物制藥有限公司 地 址:湖北省黃(huáng)梅縣小池鎮沿江路(lù)108号 證書編号:(鄂)-非經營性-2018-0070